- Première description
- Qui est atteint de polyangéite microscopique (les patients « typiques ») ?
- Symptômes classiques de la polyangéite microscopique
- Formes de vascularite similaires à la polyangéite microscopique
- Qu’est-ce qui cause la polyangéite microscopique ?
- Comment la polyangéite microscopique est-elle diagnostiquée ?
- Traitement et évolution de la polyangéite microscopique
Première description
La première description d’un patient atteint de la maladie aujourd’hui connue sous le nom de polyangéite microscopique (PMA) est apparue dans la littérature européenne dans les années 1920. Le concept de cette maladie en tant qu’affection distincte de la polyartérite noueuse (PAN) et d’autres formes de vascularite n’a cependant pas commencé à s’ancrer dans la pensée médicale avant la fin des années 1940. Aujourd’hui encore, certains termes prêtant à confusion pour l’AMP (par exemple, » poly artérite noueuse microscopique » plutôt que » poly angiite microscopique « ) persistent dans la littérature médicale. La confusion concernant la nomenclature correcte de cette maladie a conduit à des références à la « polyartérite noueuse microscopique » et à la « vascularite d’hypersensibilité » pendant de nombreuses années. En 1994, la conférence de consensus de Chapel Hill a reconnu l’AMP comme une entité à part entière, la distinguant clairement dans un schéma de classification de la PAN, de la granulomatose avec polyangéite (GPA, anciennement Wegener), de l’angiite leucocytoclasique cutanée (ALC) et d’autres maladies avec lesquelles l’AMP a été confondue au fil des ans.
L’explication de la difficulté à séparer l’AMP des autres formes de vascularite provient en grande partie des nombreuses zones de chevauchement de l’AMP avec d’autres maladies. L’AMP, la PAN, la GPA et l’ALC et d’autres troubles partagent tous une variété de caractéristiques mais possèdent suffisamment de différences pour justifier des classifications distinctes.
Qui est atteint de polyangéite microscopique ? Un patient typique
L’APM peut toucher des individus de toutes origines ethniques et de tout groupe d’âge. Aux États-Unis, le patient typique de l’AMP est un homme ou une femme blanche d’âge moyen, mais il existe de nombreuses exceptions à cette règle. La maladie peut survenir chez des personnes de tous âges, des deux sexes et de toutes origines ethniques.
Symptômes classiques de la polyangéite microscopique
De nombreux signes et symptômes sont associés à l’AMP. Cette maladie peut affecter de nombreux systèmes organiques du corps, y compris (mais pas seulement) les reins, le système nerveux (en particulier les nerfs périphériques, par opposition au cerveau ou à la moelle épinière), la peau et les poumons. En outre, les symptômes généralisés tels que la fièvre et la perte de poids sont très fréquents.
Les CINQ manifestations cliniques les plus courantes de l’AMP sont :
- Inflammation des reins (~ 80 % des patients).
- Perte de poids (> 70 %).
- Les lésions cutanées (> 60%).
- Les atteintes nerveuses (60%).
- Fièvres (55%).
Inflammation des reins
L’inflammation des reins, appelée glomérulonéphrite, entraîne une perte de sang et de protéines par les urines. Ce processus peut se produire lentement ou très rapidement au cours de la maladie. Les patients atteints d’inflammation rénale peuvent ressentir de la fatigue, un essoufflement et un gonflement des jambes.
L’image ci-dessous provient d’une analyse d’urine d’un patient atteint d’inflammation rénale. Lorsque l’AMP est active, les globules rouges forment un amas ou » plâtre » (entre parenthèses en blanc) à l’intérieur des tubules des reins enflammés. Ces « coulées » passent dans le système rénal et peuvent être observées au microscope dans l’urine du patient.
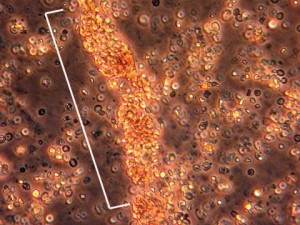
Symptômes constitutionnels
La perte de poids, les fièvres, la fatigue et le malaise font partie d’un ensemble de plaintes considérées comme des symptômes « constitutionnels ». Les plaintes constitutionnelles sont une découverte commune chez les patients atteints d’AMP, parce que la maladie est une maladie systémique se limitant généralement pas à un système organique spécifique, mais plutôt affectant largement la « constitution » du patient.
Lésions cutanées
Les lésions cutanées dans l’AMP, comme dans d’autres formes de vascularite qui impliquent la peau, peuvent faire éruption sur diverses zones du corps. Les lésions ont tendance à privilégier les zones » dépendantes » du corps, plus précisément les pieds, le bas des jambes et, chez les patients alités, les fesses. Les constatations cutanées de l’AMP cutané comprennent des bosses et des taches violacées illustrées ci-dessous (purpura palpable).
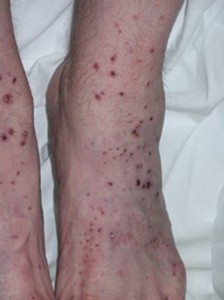
Ces zones varient en taille de plusieurs millimètres de diamètre à des lésions coalescentes encore plus grandes. Les observations cutanées dans l’AMP peuvent également inclure de petites bosses de couleur chair (papules) ; des cloques de taille petite à moyenne (lésions vésiculobulleuses) ; ou comme de petites zones de saignement sous les ongles qui ressemblent à des échardes (photo ci-dessous), d’où le nom d’hémorragies d’échardes.
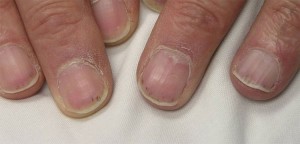
Système nerveux périphérique
Les lésions des nerfs périphériques (c’est-à-dire, les nerfs des mains et des pieds, des bras et des jambes) résultent de l’inflammation des vaisseaux sanguins qui alimentent les nerfs en nutriments. L’inflammation de ces vaisseaux sanguins prive les nerfs de leurs nutriments, ce qui entraîne un infarctus nerveux (mort des tissus). L’atteinte multiple des nerfs qui est caractéristique de la vascularite est connue sous le nom de « mononeuritis multiplex ». Cette affection est fréquemment associée à une chute du poignet ou du pied : l’incapacité d’étendre la main « en arrière » au niveau du poignet ou de fléchir le pied vers la tête au niveau de l’articulation de la cheville. Si l’affection est causée par une détérioration nerveuse associée à la vascularite, la chirurgie n’est malheureusement pas une option de traitement en raison de l’infarctus nerveux (mort des tissus).
Les symptômes neurologiques résultant de l’atteinte des nerfs périphériques peuvent également inclure un engourdissement ou des picotements dans le bras, la main, la jambe ou le pied. Avec le temps, une fonte musculaire (illustrée ci-dessous) secondaire à l’atteinte nerveuse peut résulter des dommages causés par la vascularite.
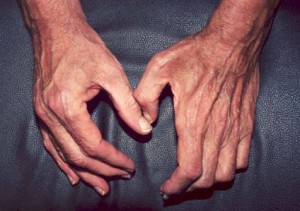
Picture:
La main de gauche (la main droite du patient) est normale, affichant un volume musculaire normal des zones entre les doigts. En revanche, la main de droite (la main gauche du patient) présente une fonte musculaire dans l’espace entre le pouce et l’index, ce qui donne à cette zone un aspect creusé, semblable à un bol. La conséquence de cette fonte musculaire est que le patient est incapable de saisir des objets entre son pouce et ses doigts (c’est-à-dire qu’il a un pincement faible) et que sa prise en main est faible.
Poumons
L’atteinte pulmonaire peut être une manifestation dramatique et potentiellement mortelle de l’AMP. Lorsque l’atteinte pulmonaire prend la forme d’une hémorragie alvéolaire, c’est-à-dire d’un saignement au niveau des petits capillaires qui sont en contact avec les sacs d’air microscopiques des poumons, l’affection peut rapidement constituer une menace pour l’état respiratoire du patient (et donc pour sa vie). L’hémorragie alvéolaire (photo ci-dessous), qui est fréquemment annoncée par l’expectoration de sang, se produit chez environ 12 % des patients atteints d’AMP .
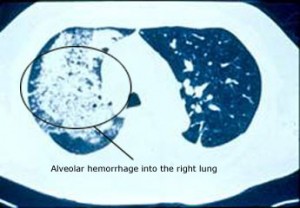
Une autre manifestation pulmonaire fréquente de l’AMP est le développement d’infiltrats inflammatoires non spécifiques, identifiables sur des radiographies de chextage ou des tomodensitométries (CT) du poumon.
Oyes, muscles et articulations
Les organes qui méritent également d’être mentionnés dans les discussions sur l’AMP sont les yeux, les muscles et les articulations. Une irritation intermittente de l’œil (ressemblant à une » conjonctivite « ) causée par une conjonctivite ou une épisclérite peut être une manifestation précoce de la maladie ou un signe de poussée de la maladie. Occasionnellement, d’autres types d’inflammation (par exemple, l’uvéite) sont également observés dans l’AMP. Les douleurs musculaires ou articulaires (connues des cliniciens sous le nom de « myalgies » ou « arthralgies », respectivement) sont des plaintes courantes dans l’AMP, accompagnant généralement les types de symptômes constitutionnels mentionnés ci-dessus. L’arthrite (inflammation des articulations accompagnée d’un gonflement) peut également être observée dans l’AMP. Les plaintes articulaires dans l’AMP et les formes de vascularite apparentées ont tendance à migrer d’une articulation à l’autre – impliquant un jour la cheville gauche, le lendemain le poignet droit, le troisième jour une épaule, par exemple.
Les formes de vascularite similaires à la polyangéite microscopique
Les similitudes et les différences entre l’AMP, la GPA et la PAN sont mises en évidence dans le tableau ci-dessous.
| MPA | GPA | PAN | |
| Vaisseau sanguin SIZE | Petite à moyenne | Petite à moyenne | Moyenne | Type de vaisseau sanguin | Artérioles à veinules, Et parfois Artères et veines | Artérioles à veinules, Et parfois des artères et des veines | Artères musculaires |
| INFLAMMATION GRANULOMATOIRE | NON | OUI | NON |
| LUNGE SYMPTOMES | OUI1 | OUI1 | NO | GLOMERULONEPHRITIS | OUI | OUI | NO | HYPERTENSION RENALE | NO | NO | YES |
| MONONEURITIS MULTIPLEX | COMMON | OCCASIONAL | COMMON | Lésions de l’écorce | YES2 | YES2 | YES2 | SYMPTÔMES DE L’IG | NO | NO | YES3 | SYMPTÔMES DE L’OEIL | YES4 | YES4 | NO | ANCA-POSITIVITÉ | 75% | 65-90% | NO |
| SYMPTÔMES CONSTITUTIONNELS | OUI5 | OUI5 | OUI5 |
| NECROTISATION DES TISSUS | OUI | OUI | OUI |
| MICROANEURYSMES | RARAREMENT | RARÉELLEMENT | TYPIQUE |
1 Capillarite pulmonaire dans l’AMP et nodules ou lésions cavitaires dans le WG
2L’AMP peut avoir des lésions cutanées de petits vaisseaux sanguins comme mentionné ci-dessus, similaires à la GPA ou des lésions des vaisseaux sanguins moyens similaires à la PAN (livedo reticularis, nodules, ulcères et gangrène digitale)
3Douleurs d’estomac après les repas
4Les complications oculaires de l’AMP sont généralement plus légères que celles de la GPA, mais de graves
problèmes oculaires, notamment une sclérite nécrosante, peuvent survenir
5Les symptômes constitutionnels comprennent une perte de poids, des fièvres, des douleurs articulaires et musculaires et des malaises.
Qu’est-ce qui cause la polyangéite microscopique ?
La cause de l’AMP n’est pas connue. Cependant, on en sait suffisamment sur quelques types de vascularites pour pouvoir décrire en termes généraux comment l’AMP affecte le corps. L’AMP est clairement un trouble médié par le système immunitaire ; les événements précis conduisant au dysfonctionnement (hyperactivité) du système immunitaire restent cependant peu clairs. De nombreux éléments du système immunitaire sont impliqués dans ce processus : neutrophiles, macrophages, lymphocytes T et B, anticorps, et beaucoup, beaucoup d’autres.
Parce que l’AMP est souvent associée à des anticorps anti-neutrophiles cytoplasmiques (ANCA), anticorps dirigés contre certains constituants des globules blancs (WBC), la maladie est souvent appelée « vascularite associée aux ANCA », ou AAV. Les ANCA, découverts en 1982, agissent contre certaines enzymes spécifiques (et naturelles) de l’organisme, présentes dans les neutrophiles et les macrophages, qui sont tous des membres de la famille des leucocytes. Le résultat des interactions des ANCA avec leurs protéines cibles est une augmentation de la destruction des GBM sur les sites de la maladie et la libération d’enzymes de globules blancs dans les parois des vaisseaux sanguins, provoquant des dommages aux vaisseaux sanguins. Dans l’AMP, les ANCA sont dirigés généralement contre des protéines spécifiques : la myéloperoxydase (MPO) et la protéinase 3 (PR3).
Comment la polyangéite microscopique est-elle diagnostiquée ?
Un prélèvement sanguin est effectué pour détecter tout taux d’ANCA, si l’AMP est suspectée. En outre, une vitesse de sédimentation des érythrocytes (VS ou » vitesse de sédimentation « ) et une protéine C-réactive (CRP) sont généralement demandées. Ces deux tests sont élevés dans de nombreux types d’inflammation différents et ne sont pas spécifiques de l’AMP ou d’une maladie particulière. L’ESR et la CRP, appelées « réactifs de phase aiguë », sont souvent des indicateurs sensibles de la présence d’une maladie active. En soi, cependant, l’élévation des réactifs de phase aiguë n’est pas suffisante pour justifier un traitement supplémentaire.
Un échantillon d’urine soigneusement analysé doit être obtenu lors de la visite initiale (et à chaque visite de suivi !) afin de rester vigilant quant au développement ou à la progression d’une atteinte rénale.
Une tomodensitométrie (TDM) du thorax peut également être réalisée pour détecter la présence d’une atteinte pulmonaire. Une biopsie tissulaire peut être nécessaire pour poser le diagnostic d’AMP, et est prélevée sur un organe qui semble être impliqué à ce moment-là. Il est parfois nécessaire de réaliser une étude d’électromyographie/conduction nerveuse (EMG/NCV) pour identifier un site pour la biopsie ou pour détecter des résultats compatibles avec une mononévrite multiplex (voir la section sur les symptômes classiques ci-dessus). Les tissus susceptibles d’être biopsiés sont le rein, la peau, le nerf, le muscle et le poumon.
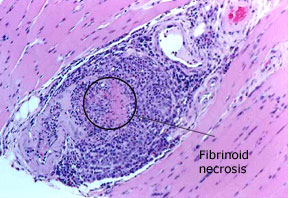
En photo : une biopsie du muscle gastrocnémien, réalisée chez un homme de 69 ans atteint de polyangéite microscopique. Un vaisseau sanguin à l’intérieur du muscle présente un infiltrat inflammatoire intense avec destruction de la paroi du vaisseau sanguin, confirmant le diagnostic de vascularite.
Traitement et évolution de la polyangéite microscopique
Un stéroïde (généralement de la prednisone) associé à un cyclophosphamide (CYC) ou au rituximab est généralement la première combinaison de médicaments à être prescrite. Après le contrôle de la maladie – généralement environ 4 à 6 mois de traitement – un traitement d’entretien sera utilisé pour maintenir la maladie en rémission. Cela varie selon les patients. La prednisone peut être interrompue après environ 6 mois.