- Primeira Descrição
- Quem apanha Poliangite Microscópica (os pacientes “típicos”)?
- Sintomas clássicos de Poliangite Microscópica
- Formas de vasculite semelhantes à Poliangite Microscópica
- O que causa a Poliangite Microscópica?
- Como é diagnosticada a Poliangite Microscópica?
- Tratamento e Curso da Poliangite Microscópica
Primeira Descrição
A primeira descrição de um doente com a doença agora conhecida como poliangite microscópica (MPA) apareceu na literatura europeia na década de 1920. O conceito desta doença como uma condição separada da poliarterite nodosa (PAN) e outras formas de vasculite só começou a enraizar-se no pensamento médico no final da década de 1940. Ainda hoje, alguns termos confusos para MPA (por exemplo, “poli arterite nodosa microscópica” em vez de “poli angite microscópica”) persistem na literatura médica. A confusão relativamente à nomenclatura apropriada desta doença levou a referências a “poliarterite nodosa microscópica” e “vasculite de hipersensibilidade” durante muitos anos. Em 1994, The Chapel Hill Consensus Conference reconheceu a MPA como entidade própria, distinguindo-a claramente num esquema de classificação da PAN, granulomatose com poliangite (GPA, anteriormente Wegener), angite leucocitocítica cutânea (ALC), e outras doenças com as quais a MPA tem sido confundida ao longo dos anos.
Muita da explicação para a dificuldade em separar a MPA de outras formas de vasculite resultou das numerosas áreas de sobreposição da MPA com outras doenças. MPA, PAN, GPA, e CLA e outras doenças partilham todas uma variedade de características mas possuem diferenças suficientes para justificar classificações separadas.
Quem apanha Poliangite Microscópica? Um doente típico
MPA pode afectar indivíduos de todas as origens étnicas e de qualquer grupo etário. Nos Estados Unidos, o paciente típico de MPA é um homem ou mulher branco de meia-idade, mas existem muitas excepções a isto. A doença pode ocorrer em pessoas de todas as idades, de ambos os sexos, e de todas as origens étnicas.
Sintomas clássicos de Poliangite Microscópica
Muitos sinais e sintomas estão associados à MPA. Esta doença pode afectar muitos dos sistemas orgânicos do corpo, incluindo (mas não só) os rins, sistema nervoso (particularmente os nervos periféricos, em oposição ao cérebro ou medula espinal), pele, e pulmões. Além disso, sintomas generalizados como febre e perda de peso são muito comuns.
As CINCO manifestações clínicas mais comuns de MPA são:
- Inflamação renal (~ 80% dos pacientes).
- Lesões da pele (> 60%).
- Lesões da pele (> 60%).
- Fevers (55%).
li>Perda de peso (> 70%).
Lesões da nervura (60%).
Inflamação renal
Inflamação nos rins, conhecida como glomerulonefrite, causa perda de sangue e proteínas através da urina. Este processo pode ocorrer lenta ou muito rapidamente no decurso da doença. Os doentes com inflamação renal podem sentir fadiga, falta de ar, e inchaço das pernas.
A imagem abaixo é de uma análise urinária de um doente com inflamação renal. Quando a MPA está activa, os glóbulos vermelhos formarão um tufo ou “gesso” (entremeado de branco) dentro dos túbulos dos rins inflamados. Estes “moldes” passam através do sistema renal e podem ser vistos ao microscópio na urina de um doente.
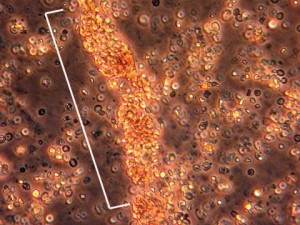
Sintomas constitucionais
Perda de peso, febres, fadiga, e mal-estar fazem parte de uma colecção de queixas consideradas como sintomas “constitucionais”. As queixas constitucionais são um achado comum em doentes com MPA, porque a desordem é uma doença sistémica que se limita geralmente não a um sistema orgânico específico, mas que afecta amplamente a “constituição” de um doente.
Lesões da pele
Lesões da pele em MPA, como noutras formas de vasculite que envolvem a pele, podem irromper em várias áreas do corpo. As lesões tendem a favorecer as áreas “dependentes” do corpo, especificamente os pés, pernas inferiores e, em pacientes acamados, as nádegas. Os achados cutâneos de MPA cutâneos incluem protuberâncias roxas e manchas retratadas abaixo (púrpura palpável).
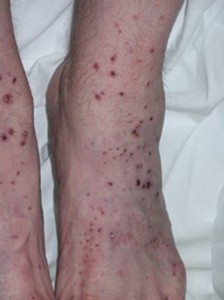
Estas áreas variam em tamanho desde vários milímetros de diâmetro até lesões coalescentes que são ainda maiores. Os achados cutâneos em MPA podem também incluir pequenas saliências da cor da pele (pápulas); bolhas de tamanho pequeno a médio (lesões vesicobolhosas); ou como pequenas áreas de hemorragia sob as unhas que parecem farpas (imagem abaixo), daí o nome de hemorragia por farpas.
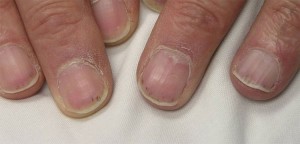
Sistema nervoso periférico
Danos aos nervos periféricos (i.e, nervos para as mãos e pés, braços e pernas) resulta da inflamação dos vasos sanguíneos que fornecem os nervos com nutrientes. A inflamação destes vasos sanguíneos priva os nervos dos seus nutrientes, levando a enfarte dos nervos (morte dos tecidos). O envolvimento múltiplo de nervos característico da vasculite é conhecido como “mononeuritis multiplex”. Esta condição está frequentemente associada à queda do pulso ou do pé: a incapacidade de estender a mão “para trás” no pulso ou de flexionar o pé para cima em direcção à cabeça na articulação do tornozelo. Se a condição for causada por deterioração nervosa associada à vasculite, infelizmente, a cirurgia não é uma opção de tratamento devido ao enfarte do nervo (morte tecidual).
Sintomas neurológicos resultantes de danos nos nervos periféricos podem também incluir dormência ou formigueiro no braço, mão, perna, ou pé. Com o tempo, o desperdício muscular (ilustrado abaixo) que é secundário ao dano nervoso pode resultar de dano causado por vasculite.
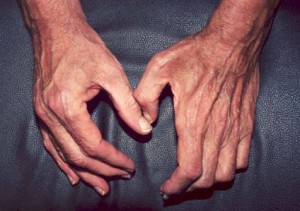
p>Pictured:
A mão à esquerda (a mão direita do paciente) é normal, exibindo massa muscular normal das áreas entre os dedos. Em contraste, a mão da direita (a esquerda do paciente) mostra o desperdício do músculo no espaço da teia entre o polegar e o primeiro dedo, levando a uma aparência oca, em forma de tigela, dessa área. A consequência deste desperdício muscular é que o paciente é incapaz de agarrar objectos entre o polegar e os dedos (ou seja, tem uma pitada fraca) e a sua mão é fraca.
Pulmões
O envolvimento pulmonar pode ser uma manifestação dramática e ameaçadora da MPA. Quando a doença pulmonar assume a forma de hemorragia alveolar – sangramento dos pequenos capilares que estão em contacto com os sacos aéreos microscópicos dos pulmões – a condição pode rapidamente representar uma ameaça para o estado respiratório do paciente (e portanto para a vida do paciente). A hemorragia alveolar (imagem abaixo), que é frequentemente anunciada pela tosse do sangue, ocorre em aproximadamente 12% dos doentes com MPA .
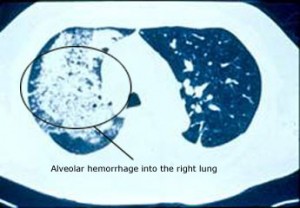
Outra manifestação pulmonar comum da MPA é o desenvolvimento de infiltrados inflamatórios não específicos, identificáveis em radiografias de queijos ou tomografia computorizada (TAC) do pulmão.
Olhos, músculos e articulações
Os órgãos que também merecem menção nas discussões de MPA incluem os olhos, músculos, e articulações. A irritação intermitente do olho (semelhante a “conjuntivite”) que é causada por conjuntivite ou episclerite pode ser uma manifestação precoce de doença ou um sinal de uma erupção de doença. Ocasionalmente, outros tipos de inflamação (por exemplo, uveíte) são também observados na MPA. As dores musculares ou articulares (conhecidas pelos clínicos como “mialgias” ou “artralgias”, respectivamente) são queixas comuns na MPA, geralmente acompanhando os tipos de sintomas constitucionais acima mencionados. A artrite (inflamação das articulações acompanhada de inchaço) também pode ser observada na MPA. As queixas articulares em MPA e formas relacionadas de vasculite tendem a migrar de uma articulação para outra – um dia envolvendo o tornozelo esquerdo, no dia seguinte o pulso direito, no terceiro dia um ombro, por exemplo.
Formas de vasculite semelhantes à Poliangite Microscópica
As semelhanças e diferenças entre MPA, GPA, e PAN são destacadas na tabela abaixo.
| >/td>>MPA | |||
| Pequeno a Médio | Medio | ||
| TIPO DE VESTILAGEM DE VENEÍCULOS A SUA VENEZA | Arteríolas a vênulas, E por vezes artérias e veias | Arteríolas a vênulas, E por vezes Artérias e veias | |
| NO | YES | NO | |
| LUNG SYMPTOMS | |||
| NO | YES | ||
| COMMON | COMMON | ||
| YES2 | YES2 | ||
| GI SYMPTOMS | NO | NO | YES3 |
| EYE SYMPTOMS | YES4 | ||
| 75% | 65-90% | NO | |
| NECROTIZAÇÃO DE TISSUE | YES | ||
| RARELY | RARELY | TYPICAL |
1 Capilarite pulmonar em MPA e nódulos ou lesões cavitárias em WG
2MPA pode ter pequenas lesões cutâneas dos vasos sanguíneos como mencionado acima, semelhantes à GPA ou lesões médias dos vasos sanguíneos semelhantes à PAN (livedo reticularis, nódulos, úlceras, e gangrena digital)
3Dores estomacais após as refeições
4MPA complicações oculares são tipicamente mais leves do que as da GPA, mas graves
problemasoculares, incluindo esclerites necrosantes podem ocorrer
5Os sintomas constitucionais incluem perda de peso, febres, dores articulares e musculares, e mal-estar.
O que causa a Poliangite Microscópica?
A causa da MPA não é conhecida. No entanto, sabe-se o suficiente sobre alguns tipos de vasculites que nos permitem descrever em termos gerais como a MPA afecta o corpo. A MPA é claramente uma desordem mediada pelo sistema imunitário; os eventos precisos que levam à disfunção do sistema imunitário (hiperactividade), no entanto, permanecem pouco claros. Muitos elementos do sistema imunitário estão envolvidos neste processo: neutrófilos, macrófagos, linfócitos T e B, anticorpos, e muitos, muitos outros.
Porque a MPA está frequentemente associada a anticorpos anti-neutrófilos citoplasmáticos (ANCA), anticorpos dirigidos contra certos constituintes dos glóbulos brancos (leucócitos), a doença é frequentemente denominada “vasculite associada a ANCA”, ou AAV. ANCA, descoberta em 1982, actua contra certas enzimas específicas (e naturais) no corpo que residem nos neutrófilos e nos macrófagos, todos eles membros da família dos leucócitos. O resultado das interacções dos ANCA com as suas proteínas alvo é um aumento da destruição dos leucócitos nos locais da doença e a libertação de enzimas dos glóbulos brancos dentro das paredes dos vasos sanguíneos, causando os danos nos vasos sanguíneos. Na MPA, os ANCA são geralmente dirigidos contra proteínas específicas: mieloperoxidase (MPO) e proteinase 3 (PR3).
Como é diagnosticada a Poliangite Microscópica?
Blood é tomado para detectar quaisquer níveis de ANCA, se houver suspeita de MPA. Além disso, é normalmente pedida uma taxa de sedimentação eritrocitária (ESR ou “sed rate”) e proteína C-reactiva (CRP). Ambos estes testes são elevados em muitos tipos diferentes de inflamação e não são específicos da MPA ou de qualquer doença em particular. O ESR e a CRP, conhecidos como “reagentes de fase aguda”, são frequentemente indicadores sensíveis da presença de doença activa. No entanto, por si só, as elevações em reactantes de fase aguda não são suficientes para justificar um tratamento adicional.
Uma amostra de urina cuidadosamente analisada deve ser obtida na visita inicial (e em cada visita de seguimento!) para manter a vigilância quer para o desenvolvimento quer para a progressão do envolvimento renal.
Uma tomografia computorizada (TAC) do tórax também pode ser realizada para detectar a presença de envolvimento pulmonar. Poderá ser necessária uma biopsia de tecido para fazer o diagnóstico de MPA, e é retirada de um órgão que parece estar envolvido na altura. Por vezes, pode ser necessário fazer um estudo de electromiografia/condução nuclear (EMG/NCV) para identificar um local para biopsia ou para detectar descobertas consistentes com um multiplex de mononeurite (ver secção de sintomas clássicos acima). Os tecidos que podem ser biopsiados são rim, pele, nervo, músculo e pulmão.
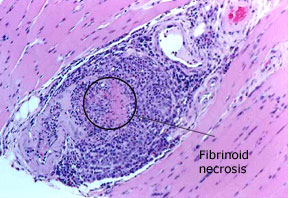
P>Pictured: uma biopsia do músculo gastrocnémico, realizada num homem de 69 anos de idade com poliangite microscópica. Um vaso sanguíneo dentro do músculo mostra um intenso infiltrado inflamatório com destruição da parede do vaso sanguíneo, confirmando o diagnóstico de vasculite.
Tratamento e Curso da Poliangite Microscópica
Um esteróide (geralmente prednisona) em combinação com uma ciclofosfamida (CYC) ou rituximab é tipicamente a primeira combinação de medicamentos a ser prescrita. Após o controlo da doença – geralmente cerca de 4 – 6 meses de terapia de manutenção do tratamento serão utilizados para manter a doença em remissão. Isto irá variar entre os pacientes. A prednisona pode ser descontinuada após aproximadamente 6 meses.