El conocimiento del metabolismo de la bilirrubina permite comprender los factores que conducen a un mayor riesgo de kernicterus (véase la imagen inferior). La bilirrubina se produce durante el catabolismo del componente hemo de los glóbulos rojos (RBC). La destrucción de los glóbulos rojos suele aumentar en el periodo neonatal inmediato; puede estar patológicamente elevada en presencia de una enfermedad hemolítica inmunomediada o no inmunomediada. La primera enzima en la cascada catabólica que conduce a la bilirrubina es la hemo oxigenasa. Se reconocen una forma constitutiva y una forma inducible que son inducidas por factores fisiológicos de estrés. La creación de bilirrubina, un compuesto insoluble en agua potencialmente tóxico, a partir de la biliverdina, una sustancia soluble en agua no tóxica, consume energía.
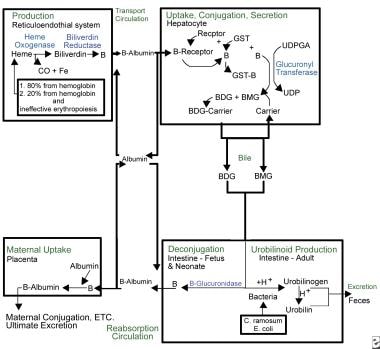 Kernicterus. Resumen del metabolismo de la bilirrubina.
Kernicterus. Resumen del metabolismo de la bilirrubina. Debido a su naturaleza lipofílica, la bilirrubina debe unirse a la albúmina para viajar por el torrente sanguíneo. En este estado, no es libre de cruzar la barrera hematoencefálica y causar kernicterus. El complejo albúmina-bilirrubina es transportado al hígado, donde la bilirrubina entra en el hepatocito para su posterior metabolismo. Una vez en el hígado, la bilirrubina se conjuga a través de la acción de la uridina difosfato glucuronil transferasa (UDPGT), una enzima que no es totalmente funcional hasta los 3-4 meses de vida.
La bilirrubina conjugada se excreta en el tracto intestinal a través del sistema biliar. La beta-glucuronidasa, presente en el lumen intestinal de los neonatos humanos, desconjuga la bilirrubina conjugada, permitiendo que se reabsorba a través de las membranas de las células lipídicas intestinales de vuelta al torrente sanguíneo, donde debe volver a unirse a la albúmina para repetir el ciclo. Este proceso, denominado recirculación enterohepática, es un fenómeno neonatal único y contribuye significativamente a la ictericia fisiológica. La alimentación y la excreción de meconio y heces interrumpen la recirculación enterohepática.
Entre los lactantes notificados en el registro de kernicterus de EE.UU., el 56% tenía anomalías que se sabe que aumentan la concentración de bilirrubina en la sangre. Se identificaron procesos hemolíticos graves en 25 de los 122 bebés (20,5%); la deficiencia de glucosa-6-fosfato deshidrogenasa se diagnosticó en 26 bebés (21,3%), el traumatismo de nacimiento se identificó en 18 pacientes (15%), y otras causas como la galactosemia, el síndrome de Crigler-Najjar y la sepsis se diagnosticaron en 8 bebés (7%). En 53 de los 122 bebés (43,4%) no se descubrió ninguna etiología para la hiperbilirrubinemia grave.
Aumento de la producción de bilirrubina
La mayor parte de la bilirrubina circulante en el neonato surge de la destrucción de los glóbulos rojos circulantes. Los neonatos producen bilirrubina a más del doble de la tasa diaria del adulto medio, principalmente debido al mayor volumen circulante de glóbulos rojos y a su menor tiempo de vida. Cualquier acontecimiento que provoque un aumento de la carga de bilirrubina sérica pone al bebé en riesgo de hiperbilirrubinemia.
Policitemia
Factores prenatales, como el tabaquismo materno, la enfermedad materna, la insuficiencia placentaria y la gestación a gran altura, pueden provocar policitemia neonatal. Los factores obstétricos, como el retraso en el pinzamiento del cordón, el desprendimiento del cordón o mantener al bebé por debajo del nivel del introito durante un periodo prolongado, pueden provocar un aumento de la masa de glóbulos rojos en el bebé. Esto es especialmente cierto en el caso de los bebés que nacen sin la presencia de una partera capacitada.
La hemólisis
La enfermedad hemolítica inmune, con mayor frecuencia la isoinmunización Rh (eritroblastosis fetal), es la etiología prototipo del kernicterus.
La isoinmunización ABO, así como los antígenos de grupos sanguíneos menores, también pueden causar enfermedad hemolítica en el recién nacido, normalmente de gravedad moderada. Los bebés nacidos de madres del grupo sanguíneo O negativo corren el mayor riesgo, y una serie de 249 bebés con hiperbilirrubinemia grave informó de una odds ratio de 48,6 para los bebés con incompatibilidad Rh.
Las anomalías del propio glóbulo rojo también pueden predisponer a la hemólisis. Pueden agruparse en defectos de membrana, como la esferocitosis hereditaria y la eliptocitosis; defectos enzimáticos, como la deficiencia de glucosa-6-fosfato deshidrogenasa y la deficiencia de piruvato quinasa; y hemoglobinopatías, como las talasemias alfa y beta.
La enfermedad de células falciformes no suele causar enfermedad hemolítica en el periodo neonatal.
La sangre extravasada
Las áreas significativas de hematomas, como el cefalohematoma grave, la hemorragia subgaleal o las equimosis periféricas del traumatismo del nacimiento, pueden dar lugar a un aumento de la carga de bilirrubina en el suero a medida que se resuelve la extracción de sangre. Las áreas internas de hemorragia, como las hemorragias pulmonares o intraventriculares, también pueden ser una importante fuente oculta de bilirrubina sérica.
Inducción de enzimas
Como se ha mencionado anteriormente, la hemo-oxigenasa-ona (HO-1) es la forma inducible de la primera enzima implicada en la creación de bilirrubina. Esta enzima se activa por factores fisiológicos de estrés, como la hipotermia, la acidosis, la hipoxia y la infección (odds ratio 20,6 en la sepsis).
Factores epidemiológicos
Los bebés del este de Asia y los nativos americanos producen bilirrubina en tasas más altas que los bebés blancos; los bebés negros tienen tasas de producción más bajas que los bebés de otros grupos raciales. Los bebés varones tienen niveles de bilirrubina sérica más altos que las mujeres. La hiperbilirrubinemia también es hereditaria; la etiología no está clara, pero puede estar relacionada con el aumento genético de los niveles de beta-glucuronidasa en el bebé, en la leche materna o en ambos (si el bebé es amamantado).
Disminución de la eliminación
Incluso con una producción normal de bilirrubina, las anomalías en el transporte, la excreción o ambas pueden dar lugar a un aumento del nivel de bilirrubina libre en el suero.
La unión de la albúmina
Debido a su naturaleza lipofílica, la bilirrubina debe unirse a una proteína portadora para ser transportada en el medio acuoso del suero. La albúmina tiene un sitio de unión primario de alta afinidad para la bilirrubina y dos sitios de menor afinidad. A pH fisiológico, la cantidad de bilirrubina libre (es decir, la bilirrubina no unida a la albúmina) es muy baja. Esto es importante porque sólo la bilirrubina libre está disponible para cruzar la barrera hematoencefálica y causar neurotoxicidad. La disminución de la capacidad de unión a la albúmina, la disminución de la afinidad de unión a la albúmina, o ambas, pueden servir para aumentar la cantidad de bilirrubina sérica libre. La afinidad de unión es menor en los neonatos que en los lactantes de más edad y es aún menor en los prematuros y en los enfermos que en los sanos a término.
Algunos autores abogan por incluir medidas de bilirrubina no ligada (es decir, libre) a la hora de evaluar el riesgo de neurotoxicidad de la bilirrubina, en parte porque algunos estudios han mostrado una asociación más estrecha entre la concentración de bilirrubina no ligada y las anomalías auditivas que las observadas con la bilirrubina sérica total, aunque sigue siendo difícil identificar el umbral de concentración de bilirrubina no ligada neurotóxica.
La disminución de la capacidad de unión puede ocurrir en la hipoalbuminemia o si los sitios de unión se llenan con otros aniones. Es controvertido si los lípidos administrados por vía parenteral pueden desplazar a la bilirrubina de su sitio de unión a la albúmina. Si se enfrentan a niveles peligrosamente altos de bilirrubina sérica, puede ser prudente restringir la administración de lípidos a niveles inferiores a los máximos. Los fármacos, como el sulfisoxazol y la ceftriaxona, también pueden competir por los sitios de unión de la bilirrubina en la molécula de albúmina y deben utilizarse con precaución o evitarse en el periodo neonatal.
Captación y conjugación hepática
La albúmina transporta la bilirrubina al hígado, donde se incorpora al hepatocito mediante una proteína aceptora llamada ligandina. Los niveles hepáticos de ligandina no alcanzan los valores adultos hasta alrededor de los 5 días de edad, pero pueden ser inducidos por la administración de fenobarbital.
Una vez dentro del hepatocito, la bilirrubina se conjuga con una fracción de azúcar, el ácido glucurónico, a través de la enzima UDPGT. La deficiencia neonatal inherente de esta enzima es la principal etiología de la ictericia fisiológica. Durante los primeros 10 días de vida, la UDPGT está presente en niveles de aproximadamente el 0,1% de los valores de los adultos, y la hiperbilirrubinemia parece ser el principal estímulo para la producción de la enzima.
Además de la ictericia fisiológica, los defectos hereditarios congénitos de la UDPGT causan hiperbilirrubinemia patológica de gravedad variable. El síndrome de Crigler-Najjar tipo I es la ausencia virtual de UDPGT y se caracteriza por una profunda hiperbilirrubinemia refractaria con el riesgo continuo de kernicterus en cualquier momento de la vida del individuo. Actualmente, el trasplante de hígado es la única terapia definitiva, aunque se están investigando terapias experimentales. Los pacientes con el síndrome de Crigler-Najjar tipo II (es decir, el síndrome de Arias) tienen una presentación clínica similar a la de los pacientes con el tipo I. Sin embargo, los pacientes con el tipo II responden dramáticamente a la terapia con fenobarbital, que es la forma en que se hace el diagnóstico.
El síndrome de Gilbert se caracteriza por una hiperbilirrubinemia crónica indirecta benigna sin evidencia de enfermedad o anormalidad hepática. La base genética de este síndrome se identificó como una repetición de tripletes amplificada en el gen codificador de UDPGT, y se sigue investigando para aclarar el posible papel del síndrome de Gilbert en los lactantes con hiperbilirrubinemia neonatal.
Excreción
Una vez conjugada, la bilirrubina hidrosoluble se excreta de forma dependiente de la energía en los canalículos biliares para su entrega final en el intestino delgado. La alteración de este sistema o la obstrucción del sistema biliar da lugar a la acumulación de bilirrubina conjugada en el suero, identificada por una elevación de la fracción directa de la bilirrubina total. La hiperbilirrubinemia directa en el neonato (definida como una fracción directa superior a un tercio de la bilirrubina total) es siempre patológica, y debe buscarse una etiología.
En el intestino delgado, la bilirrubina conjugada no puede reabsorberse. La flora intestinal la convierte en urobilinógeno, que se excreta. En el neonato, la escasez de bacterias colónicas impide esta conversión. Además, el intestino neonatal (pero no el del adulto) produce beta-glucuronidasa, una enzima que actúa sobre la bilirrubina conjugada, liberando bilirrubina libre para su posible absorción a través de la membrana lipídica de las células intestinales hacia el torrente sanguíneo. La leche materna también contiene betaglucuronidasa, y las tomas de leche materna aumentan el nivel de esta enzima en el intestino neonatal. En combinación con la lenta motilidad intestinal en los primeros días de vida, los factores anteriores dan lugar a lo que se denomina recirculación enterohepática de la bilirrubina hacia el torrente sanguíneo.
Factores sistémicos
Galactosemia
Los pacientes con este raro error congénito del metabolismo pueden presentar principalmente hiperbilirrubinemia, aunque la fracción directa suele aumentar durante la segunda semana de vida. El bebé puede manifestar otros signos característicos, como hepatomegalia, mala alimentación o letargo. La orina para sustancias reductoras, pero no para glucosa, es diagnóstica. Muchos cribados metabólicos neonatales estatales incluyen una prueba para este trastorno.
Hipotiroidismo
Aunque la etiología no está clara, la hiperbilirrubinemia indirecta prolongada es una de las características típicas del hipotiroidismo congénito, y este diagnóstico debe descartarse en cualquier bebé con hiperbilirrubinemia que persista después de las 2-3 semanas de vida. La mayoría de las pruebas metabólicas estatales incluyen un análisis de la función tiroidea, aunque los resultados falsos negativos y el retraso en la recepción de los resultados pueden hacer necesaria la realización de pruebas individuales en bebés sintomáticos.
Fármacos
La administración materna de oxitocina, diazepam o prometazina puede provocar un aumento de la bilirrubina sérica en el bebé. Del mismo modo, la administración neonatal de pancuronio e hidrato de cloral aumenta los niveles de bilirrubina. Además, algunos fármacos, como las sulfonamidas y algunas penicilinas, pueden desplazar la bilirrubina de su sitio de unión a la albúmina, aumentando efectivamente la concentración sérica de bilirrubina libre disponible para cruzar la barrera hematoencefálica.
La acidosis
La acidosis sistémica disminuye la afinidad de unión de la albúmina a la bilirrubina, dando lugar a un aumento de los niveles de bilirrubina libre en el torrente sanguíneo. La fácil disponibilidad de protones promueve la formación de ácido de bilirrubina (anión de bilirrubina libre más 2 iones de hidrógeno); esa fracción demuestra una mayor unión y transporte a las membranas de las células neuronales.
Barrera hematoencefálica alterada
La barrera hematoencefálica neonatal es más permeable a las sustancias que la del adulto. La administración de sustancias hiperosmolares, la hipercarbia, la asfixia, la infección (en particular la meningitis) y el deterioro de la autorregulación con las variaciones de la presión arterial pueden debilitar las uniones estrechas capilares, aumentando la permeabilidad capilar. Esto, a su vez, podría reducir la concentración a la que la bilirrubina es tóxica para el SNC.
Las tomas de leche materna
La ictericia fisiológica bien descrita que se observa en los primeros días de vida, especialmente en el lactante amamantado, se denomina ictericia por lactancia. Se cree que la ictericia de la lactancia es el resultado de múltiples mecanismos, descritos anteriormente, que promueven la producción e inhiben la excreción de bilirrubina, así como de la ingesta insuficiente de leche debido a la reducción de la producción de leche de las glándulas mamarias en los primeros días después del parto. La ictericia de la lactancia debe distinguirse de la ictericia de la leche materna.
Algunos lactantes amamantados, aunque clínicamente prosperan, siguen manifestando una hiperbilirrubinemia indirecta de etiología no identificable durante varios meses. Si se observa esto en un lactante amamantado, puede hacerse el diagnóstico de exclusión de ictericia por leche materna. Se cree que esta hiperbilirrubinemia está causada por niveles persistentemente altos de componentes aún no identificados en la leche materna de algunas mujeres, que dan lugar a la persistencia de la hiperbilirrubinemia del lactante. Una pista puede ser un historial de hiperbilirrubinemia similar en otros hermanos amamantados. Esta entidad es benigna.