Une bonne connaissance du métabolisme de la bilirubine permet de comprendre les facteurs conduisant à un risque accru d’ictère nucléaire (voir image ci-dessous). La bilirubine est produite lors du catabolisme du composant hémique des globules rouges (GR). La destruction des globules rouges est généralement accrue dans la période néonatale immédiate ; elle peut être pathologiquement élevée en présence d’une maladie hémolytique à médiation immunitaire ou non immunitaire. La première enzyme de la cascade catabolique conduisant à la bilirubine est l’hème oxygénase. Une forme constitutive et une forme inductible sont reconnues et sont induites par des facteurs de stress physiologiques. La création de la bilirubine, un composé insoluble dans l’eau potentiellement toxique, à partir de la biliverdine, une substance hydrosoluble non toxique, consomme de l’énergie.
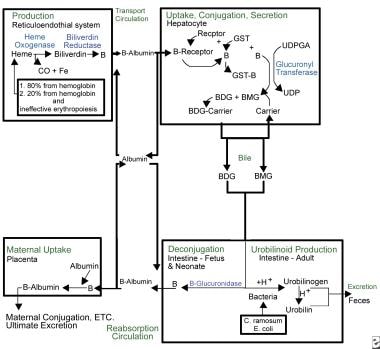 L’ictère nucléaire. Aperçu du métabolisme de la bilirubine.
L’ictère nucléaire. Aperçu du métabolisme de la bilirubine. En raison de sa nature lipophile, la bilirubine doit être liée à l’albumine pour circuler dans la circulation sanguine. Dans cet état, elle n’est pas libre de traverser la barrière hémato-encéphalique et de provoquer un ictère nucléaire. Le complexe albumine-bilirubine est transporté vers le foie, où la bilirubine pénètre dans l’hépatocyte pour y être métabolisée. Une fois dans le foie, la bilirubine est conjuguée via l’action de l’uridine diphosphate glucuronyl transférase (UDPGT), une enzyme qui n’est pas pleinement fonctionnelle avant 3-4 mois de vie.
La bilirubine conjuguée est excrétée dans le tractus intestinal via le système biliaire. La bêta-glucuronidase, présente dans la lumière intestinale des nouveau-nés humains, déconjugue la bilirubine conjuguée, ce qui lui permet d’être réabsorbée à travers les membranes des cellules lipidiques intestinales pour retourner dans la circulation sanguine où elle doit se lier à nouveau à l’albumine pour répéter le cycle. Ce processus, appelé recirculation entéro-hépatique, est un phénomène néonatal unique et contribue de manière significative à l’ictère physiologique. L’alimentation et l’excrétion du méconium et des selles interrompent la recirculation entéro-hépatique.
Parmi les nourrissons signalés dans le registre américain de l’ictère nucléaire, 56 % présentaient des anomalies connues pour augmenter la concentration de bilirubine dans le sang. Des processus hémolytiques sévères ont été identifiés chez 25 des 122 bébés (20,5 %) ; un déficit en glucose-6-phosphate déshydrogénase a été diagnostiqué chez 26 bébés (21,3 %), un traumatisme de naissance identifié chez 18 patients (15 %) et d’autres causes telles que la galactosémie, le syndrome de Crigler-Najjar et la septicémie ont été diagnostiquées chez 8 bébés (7 %). Chez 53 des 122 nourrissons (43,4%), aucune étiologie de l’hyperbilirubinémie sévère n’a été découverte.
Production accrue de bilirubine
La majeure partie de la bilirubine circulante chez le nouveau-né provient de la destruction des GR circulants. Les nouveau-nés produisent de la bilirubine à un taux quotidien plus de deux fois supérieur à celui de l’adulte moyen, principalement en raison du plus grand volume circulant de GR et de leur durée de vie plus courte. Tout événement entraînant une augmentation de la charge sérique en bilirubine expose le nourrisson à un risque d’hyperbilirubinémie.
Polycythémie
Des facteurs prénataux, tels que le tabagisme maternel, la maladie maternelle, l’insuffisance placentaire et la gestation en haute altitude, peuvent entraîner une polycythémie néonatale. Les facteurs obstétricaux, tels que le clampage tardif du cordon, le dénudage du cordon ou le maintien du bébé sous le niveau de l’introitus pendant une période prolongée, peuvent entraîner une augmentation de la masse des GR chez le bébé. Cela est particulièrement vrai pour les bébés nés en l’absence d’une accoucheuse qualifiée.
Hémolyse
Une maladie hémolytique immune, le plus souvent une isoimmunisation Rh (érythroblastose fœtale), est l’étiologie prototype de l’ictère nucléaire.
L’isoimmunisation ABO, ainsi que les antigènes de groupes sanguins mineurs, peuvent également provoquer une maladie hémolytique chez le nouveau-né, généralement de gravité modérée. Les nourrissons nés de mères de groupe sanguin O négatif sont les plus à risque, une série de 249 nourrissons atteints d’hyperbilirubinémie sévère ayant rapporté un odds ratio de 48,6 pour les nourrissons présentant une incompatibilité Rh.
Des anomalies du globule rouge lui-même peuvent également prédisposer à l’hémolyse. Elles peuvent être regroupées en anomalies membranaires, telles que la sphérocytose héréditaire et l’elliptocytose ; en anomalies enzymatiques, telles que le déficit en glucose-6-phosphate déshydrogénase et le déficit en pyruvate kinase ; et en hémoglobinopathies, telles que les thalassémies alpha et bêta.
La drépanocytose ne provoque généralement pas de maladie hémolytique pendant la période néonatale.
Sang extravasé
Les zones importantes d’ecchymoses, telles qu’un céphalhématome sévère, une hémorragie sous-galéale ou des ecchymoses périphériques dues à un traumatisme de naissance, peuvent entraîner une augmentation de la charge en bilirubine dans le sérum lorsque la collecte de sang se résorbe. Les zones internes d’hémorragie, telles que les saignements pulmonaires ou intraventriculaires, peuvent également constituer une source occulte importante de bilirubine sérique.
Induction enzymatique
Comme mentionné ci-dessus, l’hémo-oxygénase-one (HO-1) est la forme inductible de la première enzyme impliquée dans la création de la bilirubine. Cette enzyme est activée par des facteurs de stress physiologiques, tels que l’hypothermie, l’acidose, l’hypoxie et l’infection (odds ratio 20,6 dans le sepsis).
Facteurs épidémiologiques
Les bébés d’Asie de l’Est et les Amérindiens produisent de la bilirubine à des taux plus élevés que les nourrissons blancs ; les nourrissons noirs ont des taux de production plus faibles que les nourrissons des autres groupes raciaux. Les nourrissons de sexe masculin présentent des taux de bilirubine sérique plus élevés que ceux des femmes. L’hyperbilirubinémie est également présente dans les familles ; l’étiologie n’est pas claire mais pourrait être liée à une augmentation génétique des taux de bêta-glucuronidase chez le nourrisson, dans le lait maternel, ou les deux (si le nourrisson est allaité).
Décélération de l’élimination
Même en cas de production normale de bilirubine, des anomalies du transport, de l’excrétion ou des deux peuvent entraîner une augmentation du taux de bilirubine libre dans le sérum.
La liaison à l’albumine
En raison de sa nature lipophile, la bilirubine doit être liée à une protéine porteuse pour être transportée dans l’environnement aqueux du sérum. L’albumine possède un site de liaison primaire à haute affinité pour la bilirubine et deux sites à plus faible affinité. Au pH physiologique, la quantité de bilirubine libre (c’est-à-dire la bilirubine non liée à l’albumine) est très faible. Ceci est important car seule la bilirubine libre est disponible pour traverser la barrière hémato-encéphalique et provoquer une neurotoxicité. Une diminution de la capacité de liaison à l’albumine, une diminution de l’affinité de liaison à l’albumine, ou les deux, peuvent servir à augmenter la quantité de bilirubine sérique libre. L’affinité de liaison est plus faible chez les nouveau-nés que chez les nourrissons plus âgés et elle est encore plus faible chez les prématurés et les malades que chez les nourrissons à terme en bonne santé.
Certains auteurs préconisent d’inclure des mesures de la bilirubine non liée (c’est-à-dire libre) lors de l’évaluation du risque de neurotoxicité de la bilirubine, en partie parce que certaines études ont montré une association plus étroite entre la concentration de bilirubine non liée et les anomalies auditives que celles observées avec la bilirubine sérique totale, bien que l’identification du seuil de concentration de bilirubine non liée neurotoxique reste difficile à déterminer.
Une diminution de la capacité de liaison peut se produire en cas d’hypoalbuminémie ou si les sites de liaison sont remplis par d’autres anions. La question de savoir si un lipide administré par voie parentérale peut déplacer la bilirubine de son site de liaison à l’albumine est controversée. Si l’on est confronté à des niveaux dangereusement élevés de bilirubine sérique, il peut être prudent de limiter l’administration de lipides à des niveaux inférieurs aux niveaux maximaux. Les médicaments, tels que le sulfisoxazole et la ceftriaxone, peuvent également entrer en compétition pour les sites de liaison de la bilirubine sur la molécule d’albumine et doivent être utilisés avec prudence ou évités pendant la période néonatale.
Captage hépatique et conjugaison
L’albumine transporte la bilirubine vers le foie, où elle est incorporée dans l’hépatocyte par une protéine acceptrice appelée ligandine. Les niveaux hépatiques de ligandine n’atteignent pas les valeurs adultes avant l’âge de 5 jours environ, mais ils peuvent être induits par l’administration de phénobarbital.
Une fois à l’intérieur de l’hépatocyte, la bilirubine est conjuguée à une fraction de sucre, l’acide glucuronique, via l’enzyme UDPGT. La déficience néonatale inhérente de cette enzyme est la principale étiologie de l’ictère physiologique. Pendant les 10 premiers jours de vie, l’UDPGT est présente à des niveaux correspondant à environ 0,1 % des valeurs adultes, et l’hyperbilirubinémie semble être le principal stimulus de la production de l’enzyme.
Au delà de l’ictère physiologique, les défauts héréditaires congénitaux de l’UDPGT provoquent une hyperbilirubinémie pathologique de gravité variable. Le syndrome de Crigler-Najjar de type I correspond à l’absence virtuelle d’UDPGT et se caractérise par une hyperbilirubinémie réfractaire profonde avec un risque permanent d’ictère nucléaire à tout moment de la vie de l’individu. Actuellement, la transplantation hépatique est la seule thérapie définitive, bien que des thérapies expérimentales soient à l’étude. Les patients atteints du syndrome de Crigler-Najjar de type II (c’est-à-dire le syndrome d’Arias) ont une présentation clinique similaire à celle des patients atteints du type I. Cependant, les patients atteints du type II répondent de façon spectaculaire à la thérapie par le phénobarbital, ce qui est la façon dont le diagnostic est posé.
Le syndrome de Gilbert est caractérisé par une hyperbilirubinémie indirecte chronique bénigne sans preuve de maladie ou d’anomalie du foie. La base génétique de ce syndrome a été identifiée comme étant une répétition amplifiée de triplets dans le gène codant pour l’UDPGT, et les investigations se poursuivent pour clarifier le rôle possible du syndrome de Gilbert chez les nourrissons présentant une hyperbilirubinémie néonatale.
Excrétion
Une fois conjuguée, la bilirubine hydrosoluble est excrétée de manière dépendante de l’énergie dans les canalicules biliaires pour être finalement délivrée dans l’intestin grêle. Une perturbation de ce système ou une obstruction du système biliaire entraîne une accumulation de bilirubine conjuguée dans le sérum, identifiée par une élévation de la fraction directe de la bilirubine totale. L’hyperbilirubinémie directe chez le nouveau-né (définie par une fraction directe supérieure à un tiers de la bilirubine totale) est toujours pathologique, et une étiologie doit être recherchée.
Dans l’intestin grêle, la bilirubine conjuguée ne peut être réabsorbée. Les flores intestinales la transforment en urobilinogène, qui est excrété. Chez le nouveau-né, la rareté des bactéries coliques entrave cette conversion. En outre, l’intestin du nouveau-né (mais pas celui de l’adulte) produit de la bêta-glucuronidase, une enzyme qui agit sur la bilirubine conjuguée, libérant la bilirubine libre pour une absorption potentielle à travers la membrane lipidique des cellules intestinales vers la circulation sanguine. Le lait maternel contient également de la bêta-glucuronidase, et l’alimentation au lait maternel augmente le niveau de cette enzyme dans l’intestin du nouveau-né. Combinés à une motilité intestinale lente dans les premiers jours de vie, les facteurs ci-dessus entraînent ce que l’on appelle la recirculation entéro-hépatique de la bilirubine dans la circulation sanguine.
Facteurs systémiques
Galactosémie
Les patients atteints de cette erreur innée rare du métabolisme peuvent principalement présenter une hyperbilirubinémie, bien que la fraction directe augmente généralement au cours de la deuxième semaine de vie. Le bébé peut présenter d’autres signes caractéristiques, tels qu’une hépatomégalie, une mauvaise alimentation ou une léthargie. La recherche de substances réductrices dans l’urine, mais pas de glucose, permet de poser un diagnostic. De nombreux dépistages métaboliques néonatals d’État comprennent un dosage de ce trouble.
Hypothyroïdie
Bien que l’étiologie ne soit pas claire, l’hyperbilirubinémie indirecte prolongée est l’une des caractéristiques typiques de l’hypothyroïdie congénitale, et ce diagnostic doit être écarté chez tout bébé présentant une hyperbilirubinémie persistant après l’âge de 2-3 semaines. La plupart des dépistages métaboliques d’état comprennent un dosage de la fonction thyroïdienne, bien que des résultats faussement négatifs et une réception tardive des résultats puissent nécessiter des tests individuels chez les nourrissons symptomatiques.
Médicaments
L’administration maternelle d’oxytocine, de diazépam ou de prométhazine peut entraîner une augmentation de la bilirubine sérique chez le nourrisson. De même, l’administration néonatale de pancuronium et d’hydrate de chloral augmente les taux de bilirubine. De plus, certains médicaments, comme les sulfamides et certaines pénicillines, peuvent déplacer la bilirubine de son site de liaison à l’albumine, augmentant ainsi efficacement la concentration sérique de bilirubine libre disponible pour traverser la barrière hémato-encéphalique.
Acidose
L’acidose systémique diminue l’affinité de liaison de l’albumine pour la bilirubine, ce qui entraîne une augmentation des taux de bilirubine libre dans la circulation sanguine. La disponibilité immédiate de protons favorise la formation d’acide bilirubinique (anion bilirubine libre plus 2 ions hydrogène) ; cette fraction démontre une liaison et un transport accrus dans les membranes des cellules neurales.
Disruption de la barrière hémato-encéphalique
La barrière hémato-encéphalique néonatale est plus perméable aux substances que celle de l’adulte. L’administration de substances hyperosmolaires, l’hypercarbie, l’asphyxie, l’infection (notamment la méningite) et l’altération de l’autorégulation avec les variations de la pression artérielle sont autant de facteurs susceptibles d’affaiblir les jonctions serrées capillaires, augmentant ainsi la perméabilité capillaire. Ceci, à son tour, pourrait diminuer la concentration à laquelle la bilirubine est toxique pour le SNC.
Alimentation en lait maternel
L’ictère physiologique bien décrit observé dans les premiers jours de vie, en particulier chez le nourrisson allaité, est appelé ictère d’allaitement. On pense que l’ictère de l’allaitement résulte de multiples mécanismes, décrits ci-dessus, qui favorisent la production et inhibent l’excrétion de la bilirubine, ainsi que d’une consommation insuffisante de lait en raison d’une production réduite de lait de la glande mammaire dans les premiers jours du post-partum. L’ictère de l’allaitement doit être distingué de l’ictère du lait maternel.
Certains nourrissons allaités, bien que cliniquement prospères, continuent de manifester une hyperbilirubinémie indirecte d’étiologie non identifiable pendant plusieurs mois. Si ce phénomène est observé chez un nourrisson allaité, le diagnostic d’exclusion de la jaunisse due au lait maternel peut être posé. On pense que cette hyperbilirubinémie est due à la persistance de niveaux élevés de composants non encore identifiés dans le lait maternel de certaines femmes, ce qui entraîne la persistance de l’hyperbilirubinémie du nourrisson. Un indice peut être une histoire d’hyperbilirubinémie similaire chez d’autres frères et sœurs allaités. Cette entité est bénigne.
.